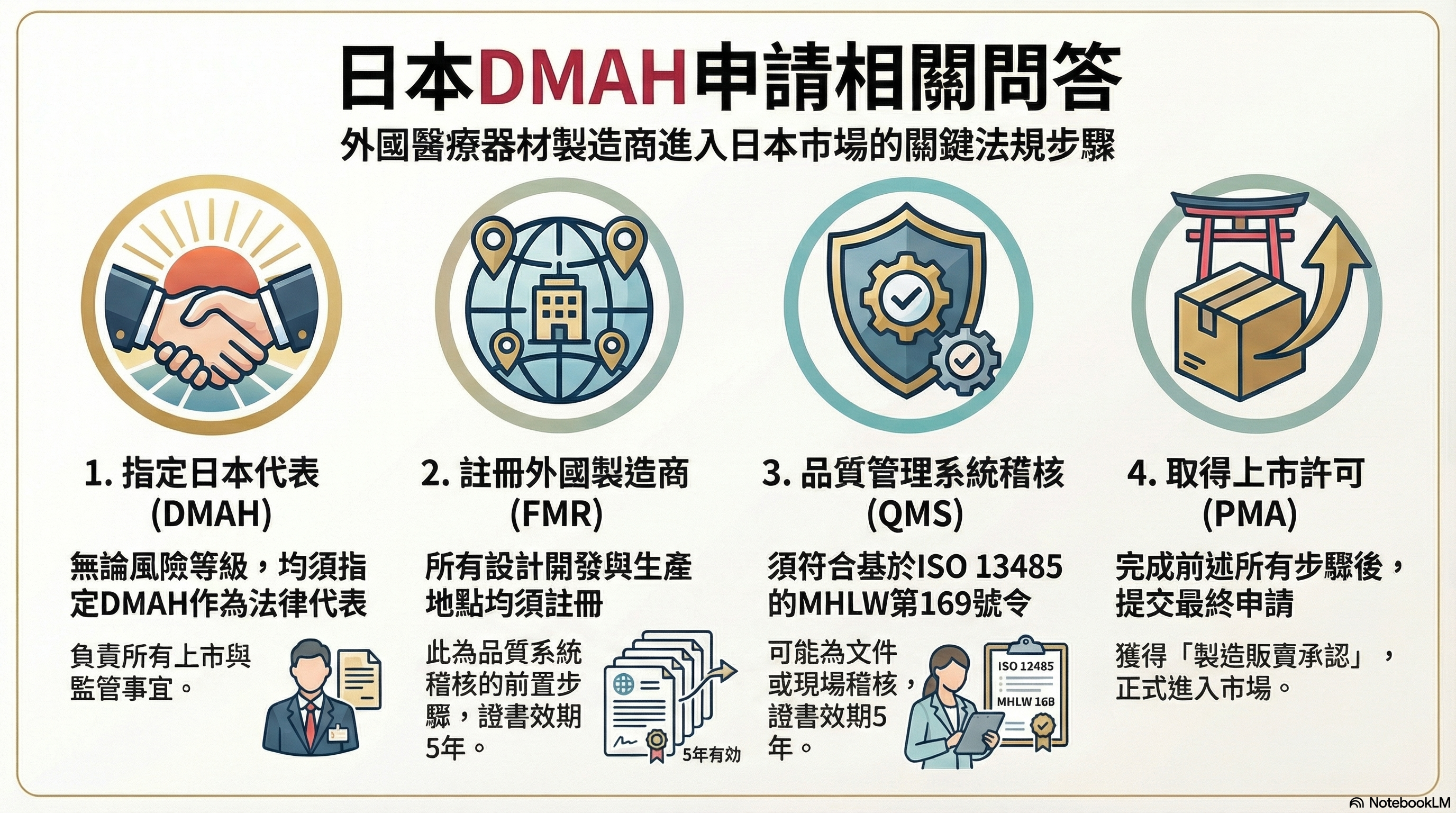
日本DMAH申請相關問答
本服務PDF介紹 Click to download PDF file
日本東京與臺灣同事攜手協同爲您服務: 安排日本DMAH (Designated Marketing Authorization Holder); 外國製造商在日本註冊登記 (FMR);品質管理系統 (MHLW MO169)稽核;上市前註冊/認證/審批 (Marketing approval,製造販売承認)。
Email:tyo4ww.mdv@evershinecpa.com
醫療器材如何銷往日本?日本醫療器材法規要求與上市途徑 (Japan’s PMD Act)為何?
日本醫療器材產品申請的法規規定主要是《藥品及醫療器材法》(PMD Act),該法於2014年11月25日生效,取代了原有的《日本藥事法》(JPAL)。
《藥品及醫療器材法》由日本厚生勞動省(MHLW)負責制定,並由醫藥品醫療機器綜合機構(PMDA)負責實施。
該法規涵蓋了醫療器材、體外診斷試劑、藥品、化妝品、可再生/細胞/基因治療等產品的上市前申請/認證/審批、製造廠註冊、日本代表(Designated Marketing Authorization Holder,DMAH)、品質管理系統、上市後安全監控等方面,以保障日本國民的安全。
如果您想要將醫療器材產品銷往日本,您需要遵守以下的程序:
• 您需要指定一個位於日本境內的DMAH,作為您在日本的法律代表。
DMAH必須持有相應的執照,並負責與日本當局溝通、提交申請文件、執行品質管理系統、處理上市後的通報等事項。
• 您需要在PMDA註冊您的製造廠,並提供相關的資料,如設計開發、生產或滅菌、工作原理等。
• 您需要根據您的產品分類分級,選擇適當的上市前審查路徑。日本將醫療器材分為四個等級,分別是Class I(低風險)、Class II(中風險)、Class III(高風險)和Class IV(最高風險)。
不同等級的產品需要提供不同程度的臨床數據和技術文件,並由不同的審查機構進行審查。
一般來說,Class I和部分Class II的產品可以通過自我認證或第三方認證機構(RCB)的審查;而Class III和部分Class II的產品則需要通過PMDA或RCB的審查。
• 您需要符合日本的品質管理系統要求,即MHLW第169號令。
該令規定了製造廠和DMAH必須建立和執行一套基於ISO 13485和GMP原則的品質管理系統,並接受PMDA或RCB的稽核。
• 您需要在上市後進行安全監控,包括收集和報告不良事件、進行上市後變更、遵守上市後警戒(Surveillance)等要求。
https://thebestra.com/japan-pmd-act/
https://origo-certification.tw/certificationshow.asp?id=5231

到日本銷售醫療器材一定要指派日本代表 (D)MAH?
無論哪一等級均須指定一位日本代表,可分為 Marketing Authorization Holder (MAH: “Seihan” in Japanese) 和 Designated Marketing Authorization Holder (DMAH: “Sennin seihan” in Japanese) 兩種。
DMAH是海外製造廠所指定日本境內的代表,類似 Authorised representative (AR,歐體代表),所有的上市許可申請、上市後警戒、通報等都要透過 DMAH 執行。(IVD、Drug 和 Medical device 的 (D)MAH 不同,各自有不同的 Licenses)
雖然日本境外製造廠須指定 DMAH 始可申請產品上市,但製造廠可透過 Foreign Exceptional Approval System (外国特例承認制度),將產品註冊在自己名下,自己成為 Foreign Restrictive Authorization Holder (FRAH),而非 DMAH 名下。
DMAH 由已取得產品上市許可的境外製造廠指定位於日本境內的 MAH 作為其 DMAH。
境外製造廠為上市前認證/審批的申請者,也是產品上市許可的擁有者。
DMAH 在申請上市認證/審批間 / 後,被境外製造廠授權代為執行相關作業。
須獲得境外製造廠的授權,DMAH 始可轉換產品上市許可。
Regardless of the classification level, a Japanese representative must be designated, and there are two types: Marketing Authorization Holder (MAH), known as “Seihan” in Japanese, and Designated Marketing Authorization Holder (DMAH), known as “Sennin seihan” in Japanese.
DMAH serves as the representative of overseas manufacturers within Japan, similar to an Authorized Representative (AR) in the European Union. All applications for marketing authorization, post-marketing surveillance, and reporting are carried out through the DMAH. (Note that IVD, drug, and medical device DMAHs have different licenses.)
While Japanese overseas manufacturers are required to designate a DMAH for product marketing applications, manufacturers can utilize the Foreign Exceptional Approval System to register the product under their own name, becoming a Foreign Restrictive Authorization Holder (FRAH) instead of a DMAH.
DMAH is designated by overseas manufacturers that have obtained product marketing authorization to appoint a Japanese MAH as their DMAH within Japan. The overseas manufacturer acts as the applicant for pre-market certification/approval and is the holder of the product marketing authorization.
DMAH is authorized by the overseas manufacturer to carry out relevant operations during or after the application for marketing certification/approval.
Authorization from the overseas manufacturer is required for DMAH to transfer the product marketing authorization.
外國製造商註冊登記 (FMR),又稱為上市前註冊(Pre-Market Submission, PMS)?
外國製造商註冊登記 (FMR) 屬於上市前註冊(Pre-Market Submission, PMS).
原本以為要先品質管理系統稽核後,再進行 FMR。但〈QMS regulation in Japan〉(PMDA, 2015) 寫:「Manufacturer needs “Registration” before the QMS inspection is conducted. 」
所以,FMR 應該比較像台灣的工廠登記。
無論等級的外國醫材製造商,所有負責設計開發 (Development) 、製造 (Production)、最終組裝 (Final assembly) 的地點,均須由 DMAH 向 MHLW 遞交 Form 63-5 申請 Foreign Manufacturer Regisration (FMR),成為 Accredited Foreign Manufacturer,此流程稱為「Gaikoku seizo-gyosya nintei (外国製造業者認定)」。
FMR 證書效期為 5 年,MHLW 建議於到期日前 5 個月開始更新證書。
Foreign Manufacturer Registration (FMR) is part of the Pre-Market Submission (PMS) process.
Initially, it was believed that a Quality Management System (QMS) audit should precede FMR. However, according to the QMS regulation in Japan (PMDA, 2015), it states: “Manufacturer needs ‘Registration’ before the QMS inspection is conducted.”
Therefore, FMR can be likened to factory registration in Taiwan. Regardless of the classification level, all foreign medical device manufacturers responsible for design development, production, and final assembly locations must submit Form 63-5 through DMAH to MHLW to apply for Foreign Manufacturer Registration (FMR) and become an Accredited Foreign Manufacturer. This process is referred to as “Gaikoku seizo-gyosya nintei” in Japanese.
The FMR certificate is valid for 5 years, and MHLW recommends starting the renewal process 5 months before the expiration date.
品質管理系統 (MHLW MO169)稽核,上市前認證(Pre-Market Certification, PMC)?
品質管理系統 (MHLW MO169)稽核屬於上市前認證(Pre-market certification, PMC)
要先進行 FMR後,再品質管理系統稽核。
所有等級的醫材製造商均須遵循 MHLW MO169 (Ordinance #169),此第169号法令是基於 ISO 13485 所建立的。在 2021 年 3 月 26 日 MO169 為了調和 ISO13485:2016 而改版,轉換期為 3 年,因此 DMAH 最遲須在 2024 年 3 月 25 日前符合此版本的 MO169。(版本差異請參考 PMDA「Revision of Japanese Medical Device QMS requirements」)
審查大約耗時 4 個月。
MO169 第三章節針對 (D)MAH 額外規定 QMS 文件/紀錄的保存期限、不良事件通報、優良警示規範 (Good Vigilance Practice,GVP) 等等。(細節請參考 PMDA「Tentative translation of MHLW MO 169 Chapter 3, as revised in 2021」)
大部分 Class II 醫材 (Specified Controlled) 的製造商品質管理系統 (QMS) 由 RCB 稽核;部分 Class II (Controlled) 以及 Class III、Class IV 製造商的 QMS 則須由 PMDA 稽核。
上述稽核均是在核發產品上市許可前稽核 DMAH 以及 Accredited Foreign Manufacturer,可能透過現場稽核或文件稽核,視是否有 ISO 13485、通報事件、召回,或依製程複雜度而定。
PMDA 一般對境外製造廠大多為文件稽核 (Document audit)。文件稽核時會確認下列資料:
- 製造現場總覽
- 組織架構
- 品質手冊
- 品質文件列表
- 製造流程 (含確效)
- MAH 合約
- 上市後警戒程序
若製造廠已有 Medical Device Single Audit Program (MDSAP) 稽核報告,一般來說,可免於重覆稽核的問題。
QMS 證書效期 5 年,且須在到期前 6 個月開始更新。
Regarding the Quality Management System (MHLW MO169) audit:
The MHLW MO169 audit is part of the Pre-Market Certification (PMC) process.
FMR must be completed before conducting the Quality Management System audit.
All medical device manufacturers, regardless of classification level, must adhere to MHLW MO169 (Ordinance #169), which is based on ISO 13485. On March 26, 2021, MO169 was revised to harmonize with ISO 13485:2016, with a transition period of 3 years. Therefore, DMAH must comply with this version of MO169 by March 25, 2024, at the latest. (For version differences, please refer to PMDA’s “Revision of Japanese Medical Device QMS requirements.”)
Chapter 3 of MO169 includes additional requirements for QMS documentation/record retention, adverse event reporting, Good Vigilance Practice (GVP), and more, specifically for (D)MAH. (For details, please refer to PMDA’s “Tentative translation of MHLW MO 169 Chapter 3, as revised in 2021.”)Most Class II medical devices (Specified Controlled) have their QMS audited by RCB, while some Class II (Controlled) and Class III, Class IV manufacturers require audits by PMDA.
These audits are conducted before issuing product marketing approval and may involve on-site audits or document audits, depending on factors such as ISO 13485 compliance, incident reporting, recalls, or the complexity of the manufacturing process.
PMDA generally conducts document audits for overseas manufacturers, verifying the following information during the audit:
Overview of manufacturing facilities
Organizational structure
Quality manual
List of quality documents
Manufacturing processes (including validation)
MAH contracts
Post-marketing surveillance procedures
If the manufacturing facility already has a Medical Device Single Audit Program (MDSAP) audit report, it can generally be exempt from duplicate audits.
QMS certificates are valid for 5 years and should be renewed starting 6 months before the expiration date.
上市前註冊/認證/審批,製造販売承認(Pre-Marketing Approval, PMA)?
上市前審批(Pre-Market Approval, PMA)
DMAH 由已取得產品上市許可的境外製造廠指定位於日本境內的 MAH 作為其 DMAH。
境外製造廠為上市前認證/審批的申請者,也是產品上市許可的擁有者。
DMAH 在申請上市認證/審批間 / 後,被境外製造廠授權代為執行相關作業。
須獲得境外製造廠的授權,DMAH 始可轉換產品上市許可。
審查大約耗時 12 個月。
Pre-Marketing Approval (PMA) refers to the process that a product must undergo before being marketed.
Pre-Market Approval (PMA)
DMAH is a Designated Marketing Authorization Holder appointed by an overseas manufacturer that has obtained product marketing authorization for their product in Japan.
The overseas manufacturer is also the applicant for pre-market certification/approval and holds the product’s marketing authorization.
DMAH, with the authorization from the overseas manufacturer, carries out relevant operations either before or after applying for marketing certification/approval.
Authorization from the overseas manufacturer is necessary for DMAH to transfer the product’s marketing authorization.
Contact us:
Email:tyo4ww.mdv@evershinecpa.com
或
日本永輝BPO有限公司 -Evershine.jp
Onarimon Yusen Building 7F,Nishi-Shinbashi 3-23-5,Minato-ku, Tokyo 105-0003, Japan
丘玲恵(Kyu Reike)日本籍来自台灣
神內伶靜(Jinnai Rinsei)日本永居来自福建
或
朱鑑彰 Jerry Chu 協理
手機 and Whats App:+886-939-357-735
WeChat: jc0096
電話:+886-2-2717-0515 分機:103
附加資訊
永輝100%關係企業
永輝總部、得恩康人力資源、永輝智權、臺北永輝、廈門永輝、北京永輝、上海那靈、深圳常新、紐約永輝、加州永輝、德州永輝、鳳凰城永輝、、首爾永輝、河內永輝、越南胡志明、曼谷永輝、新加坡永輝、吉隆玻永輝、雅加達永輝、馬尼拉永輝、墨爾本永輝、澳洲雪梨、孟加拉永輝、新德里永輝、印度孟買、杜拜永輝、法蘭克福永輝、巴黎永輝、倫敦永輝、荷蘭永輝、西班牙永輝、義大利永輝、羅馬尼亞永輝、多倫多永輝、墨西哥永輝。
其他已提供中文化服務城市:
邁阿密、亞特蘭大、俄克拉荷馬、密歇根、西雅圖、特拉華;
柏林; 斯圖加特;布拉格;布加勒斯特;班加羅爾;泗水;
高雄、香港、深圳、東關、廣州、清遠、永康、杭州、蘇州、崑山、南京、重慶、許昌、青島、天津。
永輝潛在可服務城市 (2個月籌備期):
我們為IAPA會員所,總部在倫敦,全球300個會員所,員工約1萬人。
我們為LEA會員所,總部在美國芝加哥,全球600個會員所,員工約2萬8千人。
Evershine is local Partner of ADP Streamline® in Taiwan.
(版本:2022/03)
更多城市更多服務 請點擊 網站導覽



